Computational RNA Biology
Wastewater Metagenomics
Emanuel Wyler, Markus Landthaler and I are developing computational pipelines for analyzing and monitoring wastewater data across Berlin and other environmental sites. See Wastewater pipeline here.
We are trying to understand the taxonomic complexity of these comprehensive pools of organisms and RNA where we are aiming to discover new sequences (such as new variants of CRISPR-Cas proteins) within the wastewater to be used as novel substances for developing novel biotechnologies.
Preprint: Comprehensive profiling of wastewater viromes by genomic sequencing


Codon Bias of mRNA
Markus Landthaler and I are investigating the Codon usage bias of mRNA associated with Endoplasmic Reticulum (ER). Specifically, we are interested in certain sites of mRNA that interacts with HDLBP, a RNA-binding protein that regulates the translation of mRNA around ER. Currently, we are investigating the codon usage of mRNA that are interacting with HDLBP, specifically pairs of codons that are abundantly used by these binding sites as well as their precise locations. See CodonUtil package here.


Single Cell Analysis
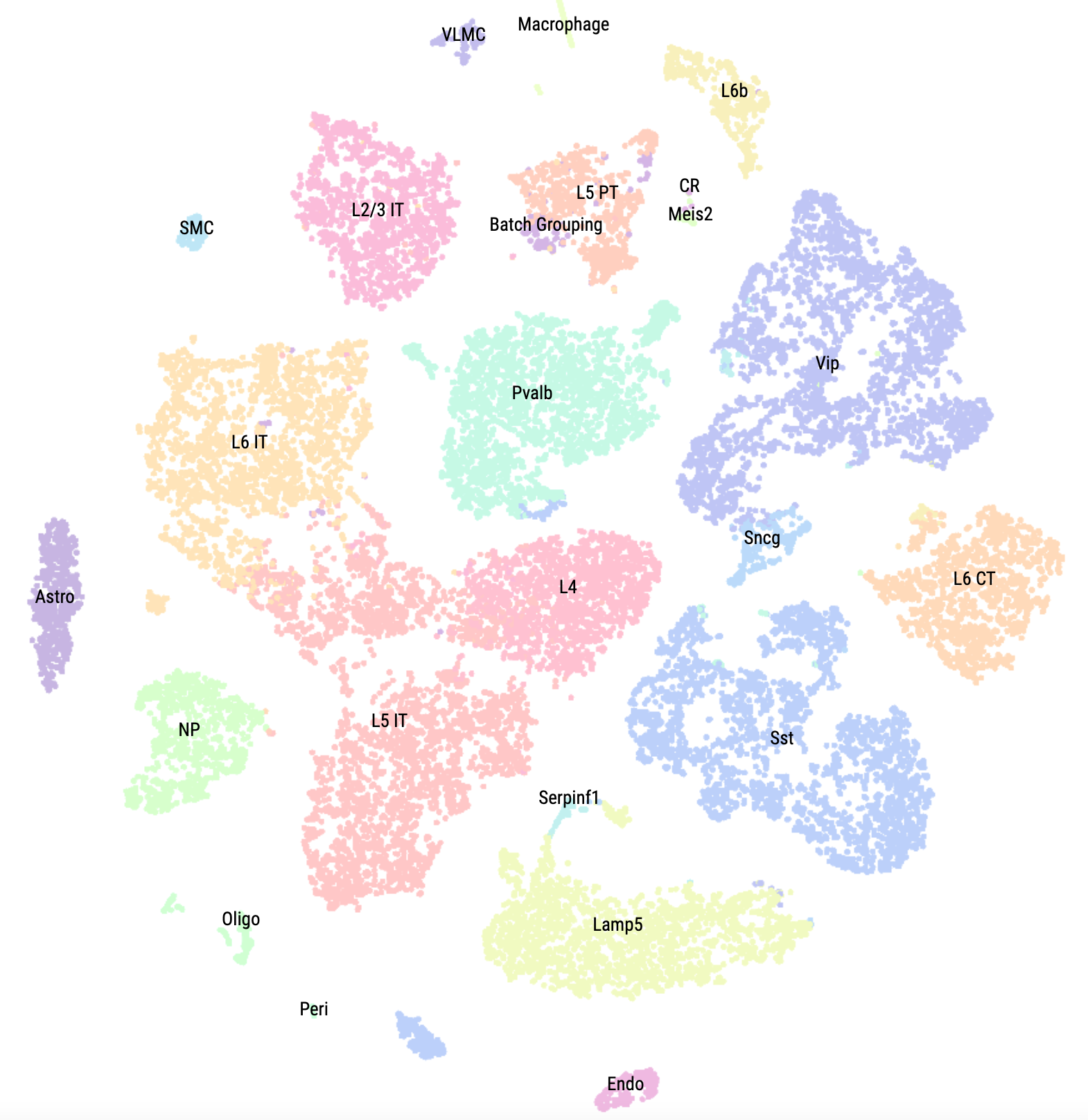
Multiomic Brain Atlas
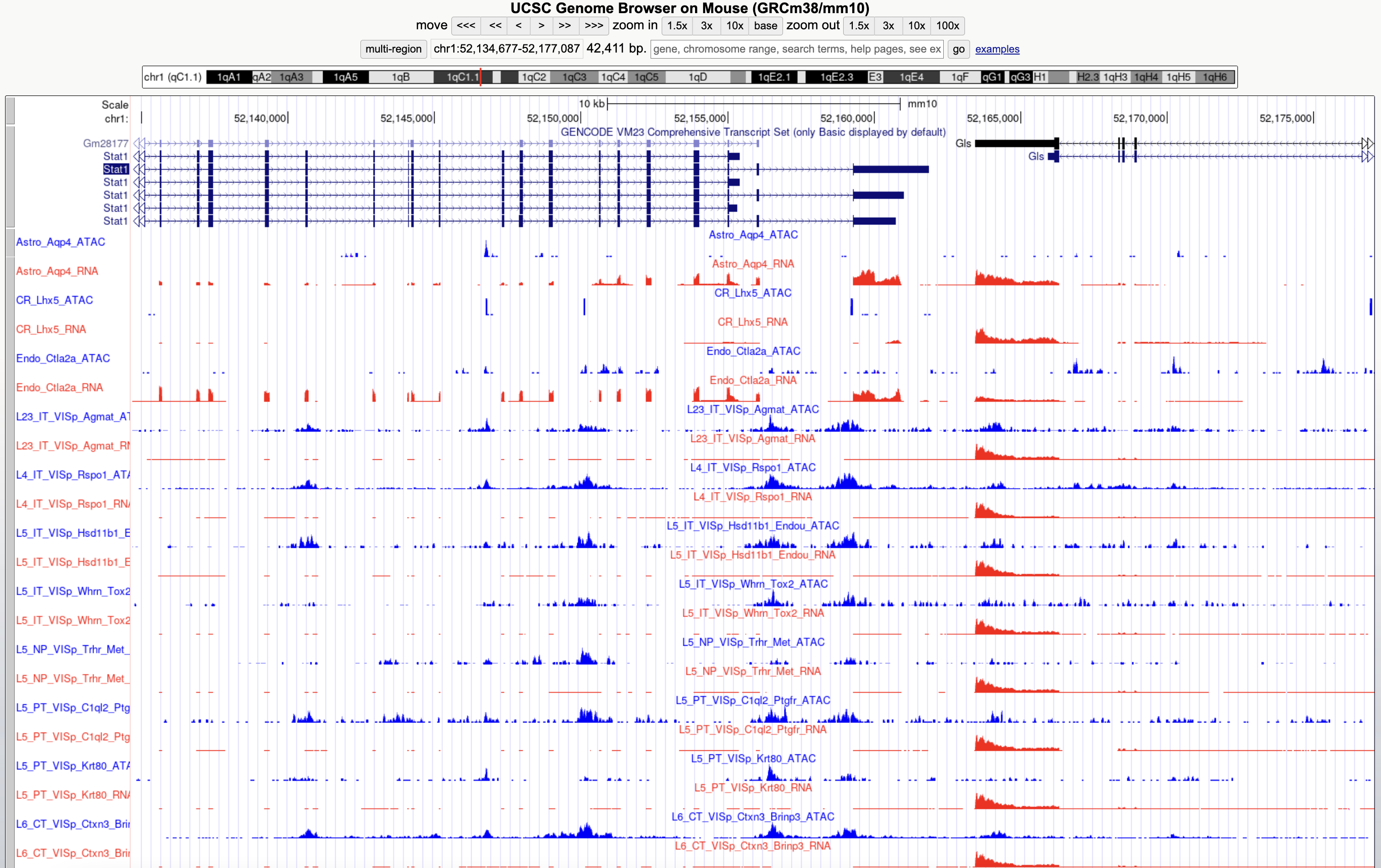
This single cell brain atlas provides access to processed and analyzed data of human and mouse single cell (scRNA-Seq and scATAC-Seq) from large single cell multiomics studies conducted in Allen Institute for Brain Science as well as samples from additional projects such as FANTOM5 for CAGE-Seq. This integrative project has been conducted in collaboration with https://www.umassmed.edu/gtc/about/faculty-publications/miguel-sena-esteves/ and at UMass Chan Medical School.
In collaboration with Sena-Esteves Lab and Bioinformatics Core at UMass Chan Medical School, this website hosts a browser for interactively visualizing transcriptional profiles of mouse brain cell types, and two publicly accessible custom track hubs (for human and mouse) within UCSC genome browsers.
Website: https://neocortex.dolphinnext.com/
Single Cell Data (Cellxgene): https://neocortex.dolphinnext.com/cellxgenebrowser.html
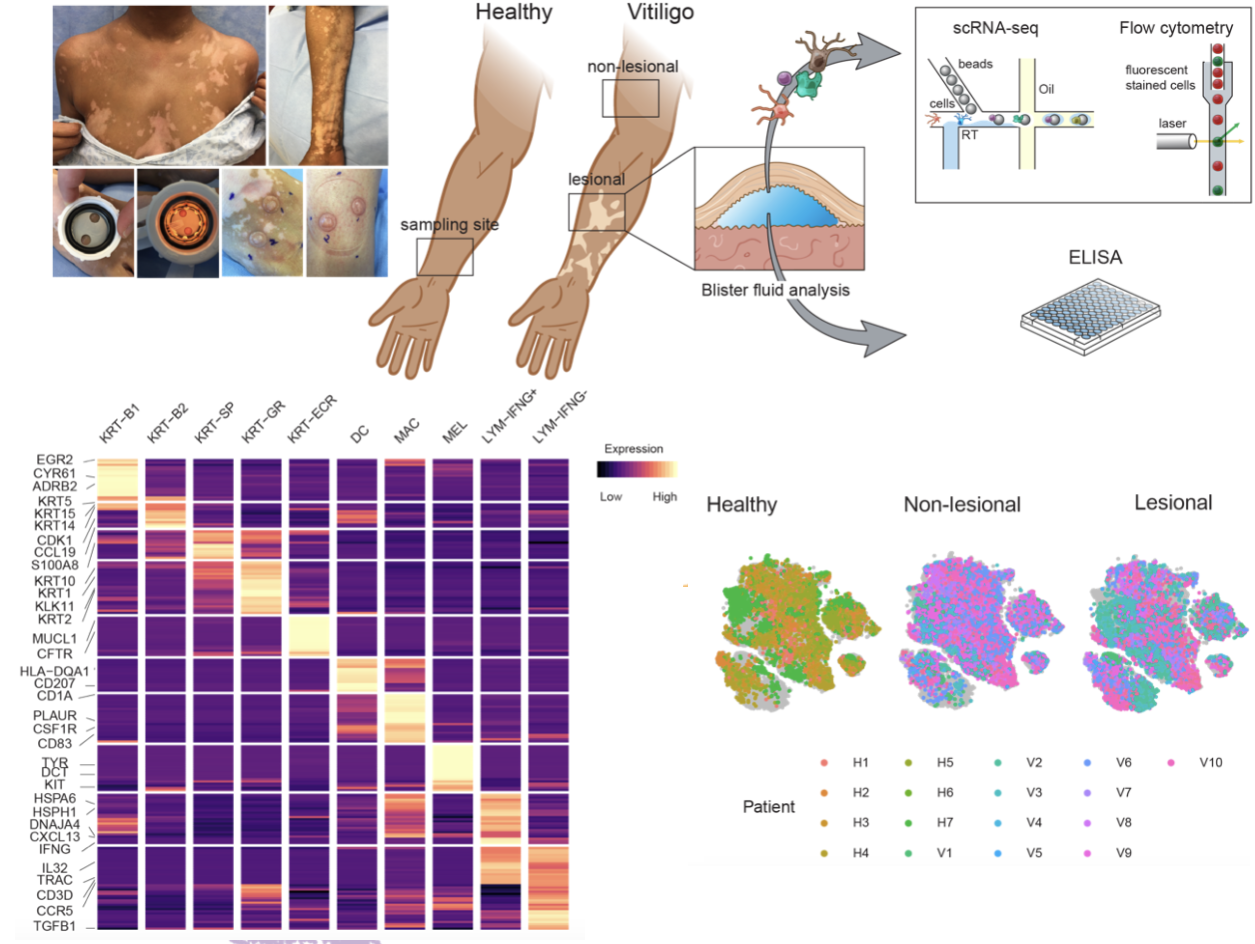
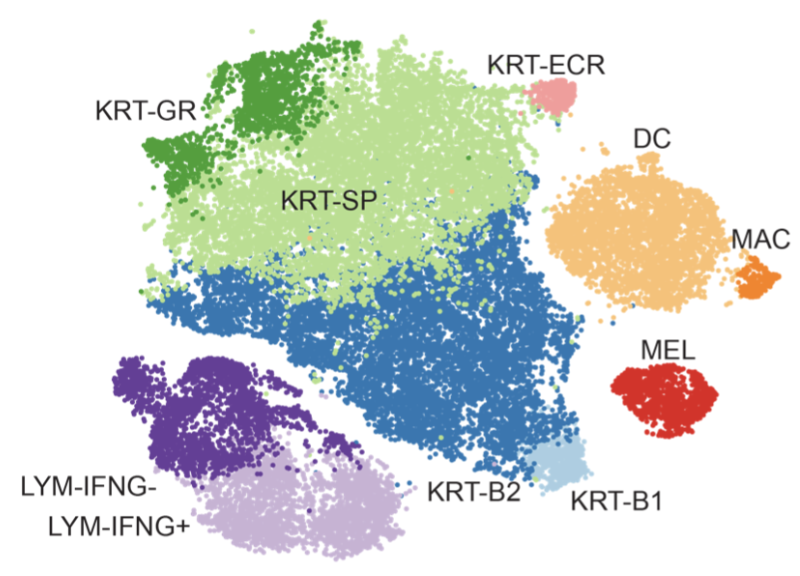
Vitiligo Atlas
In collaboration with Garber Lab and Bioinformatics Core at UMass Chan Medical School, we provided access to the data we used in our recent publication (Gellatly et. al 2021) where we sought to identify cellular communications that show disruption in lesions or in non-lesional skins of individuals suffering from Vitiligo, an autoimmune skin disease characterized by the targeted destruction of melanocytes by T cells.
The data provided was obtained using our in-house inDrop system to generate single cell RNA-Seq profiles on affected and unaffected skin from vitiligo patients, as well as healthy controls. The data was analyzed using an end-to-end scRNA data analysis package called SignallingSingleCell.
All raw sequencing data was processed by the scRNA-Seq inDrop pipeline developed within the interactive pipeline manager DolphinNext (Yukselen et. al 2020).
Website: https://vitiligo.dolphinnext.com/
Single Cell Data (Cellxgene): https://vitiligo.dolphinnext.com/browse.html